Description
AA sequence: ECRYLFGGCSSTSDCCKHLSCRSDWKYCAWDGTFS-OH
Disulfide bonds: Cys2-Cys16, Cys9-Cys21, Cys15-Cys28
Length (aa): 35
Formula: C170H239N47O54S6
Molecular Weight: 3997.46 g/mol
CAS number:
Source: Synthetic
Purity rate: > 95 %

270 € – 385 €
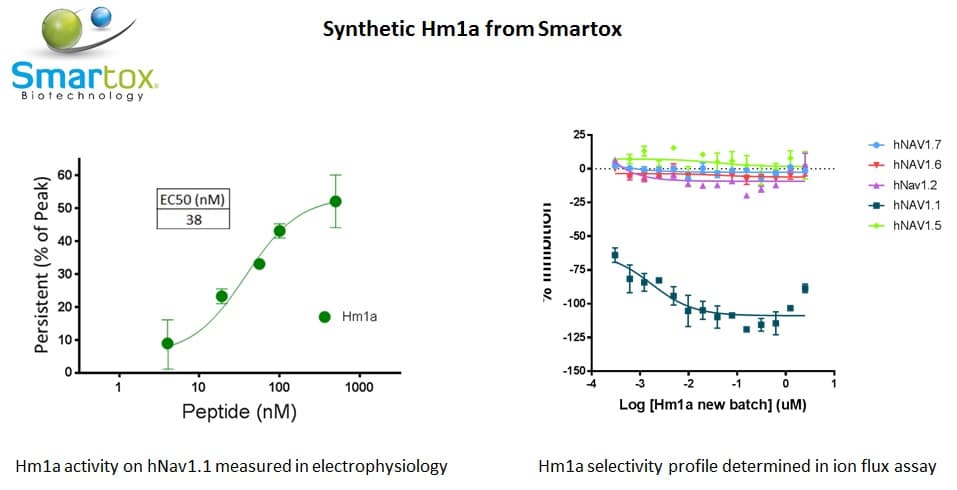
Hm1a has been identified from the venom of the spider Heteroscodra maculata. Hm1a has been described as a potent and selective agonist of the voltage-gated sodium channel Nav1.1 with an EC value of 38 nM. Nav1.1 is expressed in the CNS and mutations are associated with several disorders such as epilespy or autism.
Smartox Biotechnology is pleased to offer a synthetic and functionally active Hm1a dedicated to research laboratory use only.
AA sequence: ECRYLFGGCSSTSDCCKHLSCRSDWKYCAWDGTFS-OH
Disulfide bonds: Cys2-Cys16, Cys9-Cys21, Cys15-Cys28
Length (aa): 35
Formula: C170H239N47O54S6
Molecular Weight: 3997.46 g/mol
CAS number:
Source: Synthetic
Purity rate: > 95 %
